
























 Литература
ЛитератураФилософ и художник: Яков Друскин и Михаил Шемякин
 Была ли крокоутка?
Была ли крокоутка?
 Чем занимаются генетики?
Чем занимаются генетики?
 Кто на Луне главный?
Кто на Луне главный?
 Как возникает лжеистория?
Как возникает лжеистория?
 Что делают медики на войне?
Что делают медики на войне?
 Как рассказать о блокаде Ленинграда?
Как рассказать о блокаде Ленинграда?
 Что придумал Толкин?
Что придумал Толкин?
 Как возникла Вселенная и что с ней будет дальше?
Как возникла Вселенная и что с ней будет дальше?
 Как «Радио Свобода» сохранило запрещенную литературу
Как «Радио Свобода» сохранило запрещенную литературу
 Как спорт превратился в шоу-бизнес?
Как спорт превратился в шоу-бизнес?
 Почему мы такие умные?
Почему мы такие умные?
 Роботы и музыка
Роботы и музыка
 © Политехнический музей
© Политехнический музейВ Лектории Политехнического музея заведующий лабораторией функциональной геномики Медико-генетического научного центра Михаил Скоблов рассказал, к чему в итоге приведут исследования в области медицинской генетики и почему первый «отредактированный» человек, скорее всего, родится в Китае.
Начну с того, что коротко расскажу о себе. Я больше десяти лет работаю в Медико-генетическом научном центре, который занимается генетическими заболеваниями человека — тем, как они устроены, каковы их причины — и разрабатывает разные подходы для их диагностики и лечения. Также я работаю в Московском физико-техническом институте, где созданы хорошие условия для занятий наукой. В МФТИ меня в первую очередь интересуют студенты — талантливые ребята, которые уже сейчас могут стать участниками научного процесса, в частности, помогать обрабатывать огромное количество данных, продолжающих накапливаться в области биологии и медицинской генетики.
А что же это такое — медицинская генетика? Одно из простых объяснений: это наука, занимающаяся выяснением роли генов в возникновении патологий у человека. Как известно из школьного курса, вся генетика пошла от Менделя (Грегор Иоганн Мендель — австрийский ботаник, монах-августинец, основоположник учения о наследственности. — Ред.); так вот, те же самые классические законы наследования, им прописанные, лежат и в основе современной медицинской генетики.
Начнем с наследственных болезней. Я расскажу, как их находили, как описывали, как изучали. Вообще это довольно сложный вопрос. Вычленить генетические заболевания долгое время не удавалось. Это совсем не простая задача. Но основным краеугольным камнем в медицинской генетике является понимание наследования заболеваний. И в основе этого лежит так называемая родословная семьи больного.

Квадратиками в таких «родословных» всегда обозначаются лица мужского пола, кружочками — лица женского пола. Вот у них образуются дети, которые тоже могут давать потомство, и так далее. И вот в каком-то поколении возникает один больной член семьи, и понятно, что его болезнь может как-то наследоваться. И тут возникает та самая генетическая компонента, которую можно вычленить, расписать, с ней медицинская генетика уже может начинать работать.
Самое первое вычленение генетической компоненты случилось относительно недавно — в 1966 году. Был такой ученый Виктор Алмон Маккьюсик, который создал каталог аутосомно-доминантных, аутосомно-рецессивных и Х-сцепленных фенотипов (то есть то, как люди выглядят, как в них проявляются заболевания). И с тех пор весь мир занимается исследованиями и сбором информации о том, как выглядят генетические заболевания, как они устроены. На сегодняшний день их описано очень много — больше восьми тысяч. Существует онлайн-база данных OMIM (Online Mendelian Inheritance in Man), в которой любой ученый, если он провел грамотное исследование, может оставить запись, тем самым обогатив науку.

Какие-то заболевания известны очень хорошо — мы знаем их молекулярные основы и понимаем, из чего они происходят. Для каких-то вcе еще не хватает информации. А про какие-то болезни пока только предполагается, что они могут быть генетическими. Но это самая важная основа медицинской генетики: мы имеем описания заболеваний, которые на сегодняшний день удалось сделать, и их теперь можно исследовать.
В целом моногенные болезни — то есть когда поломки в каком-то гене приводят к заболеванию — проявляются в раннем детском возрасте. Большая их часть — почти 90% — диагностируются в младенчестве. Менее 10% проявляется после полового созревания и только 1% — в конце репродуктивного периода. Логика понятна: если происходит какая-то поломка в генетическом материале, перестает функционировать какой-то белок, то, как правило, проявления этой поломки видны с первых дней жизни, а очень часто даже внутриутробно. Но если все-все заболевания сложить, то — в случае моногенных заболеваний, когда поломка в одном гене приводит к одному заболеванию, — частота их проявлений составляет 0,36%. Скажу иначе: из тысячи человек только четырем грозит быть обладателями генетического заболевания. Но все эти болезни исследуются самым подробным образом. Что это за болезни? В России наиболее часто встречающимися являются следующие:

Для муковисцидоза: один больной встречается на восемь тысяч человек. Для фенилкетонурии: один на десять тысяч. То есть самое частое заболевание — оно же и редкое. Но суммарно мы имеем достаточно большую цифру.
Чтобы двигаться дальше, хочу коротко объяснить, как такие заболевания вообще возникают. Существует два механизма, два варианта наследования: аутосомно-рецессивный и аутосомно-доминантный. Чем они отличаются?

У каждого из нас есть два набора хромосом: одни пришли от папы, другие от мамы. В случае аутосомно-рецессивного заболевания каждый из родителей может нести поломки гена в одной хромосоме — одна поломка в маминой, другая в папиной. Соответственно, когда у них рождаются дети, то возможны три варианта: рождается больной ребенок, у которого поломки двух копий генов, рождается двое детей, и у каждого по одной поломке, или ребенок, у которого нет ни одной поломанной копии гена. То есть в случае аутосомно-рецессивного заболевания в потомстве только один ребенок может иметь обе копии поломанных генов, в результате чего и возникает заболевание. Считается, что в среднем в популяции каждый из нас может быть носителем восьми или даже десяти мутантных аллелей (то есть разных форм одного и того же гена). То есть мы можем (не дай бог, конечно) встретить партнера, у которого будет поломка в том же самом гене, и это приведет к тому, что будут рождаться дети по вот такому распределению. Аутосомно-рецессивный тип наследования встречается почти у половины всех генетических заболеваний человека.

Второй вариант — аутосомно-доминантный тип наследования. Здесь картина еще проще: достаточно поломки лишь в одной копии гена, и возникает заболевание. Поэтому если мы имеем одного больного родителя, то это заболевание будет четко наследоваться с 50-процентной вероятностью его детьми. Почему здесь одно наследование, а там другое? Ну, вот так устроены гены. Иногда ген настолько важный, что его недостаток приводит к тому, что заболевание возникает. А бывает, что у детей или у самих родителей половина копий генов нормальная, половина с мутацией, но срабатывает компенсаторный механизм, который помогает организму с этим справляться, и заболевание никак не развивается.
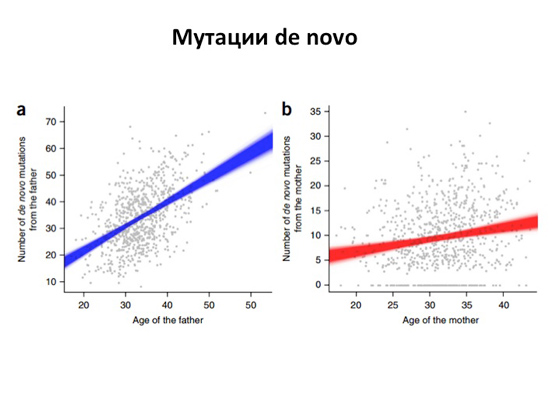
Откуда берутся эти самые поломанные гены с этими самыми мутациями? Понятно, что мы имеем в своих клетках ДНК, и ДНК эта очень-очень большая. Три миллиарда нуклеотидов! Соответственно, когда клетки делятся, как бы ни был точен процесс деления, возникают ошибки. Несмотря на то что у нас в клетках существуют механизмы, которые смотрят за тем, чтобы ошибок не было, и даже репарируют неправильные замены, — все равно какая-то часть мутаций возникает и наследуется. И долгое время было непонятно: а как же часто это происходит, как это все устроено? И лишь недавно — буквально в последние несколько лет — вышло несколько очень мощных научных работ. Геномы здоровых и больных людей полностью секвенировали, чтобы возможно было оценить, как возникают новые мутации относительно первого поколения и второго, и оказалось, что в среднем от отца дети наследуют около сорока каких-то новых изменений. То есть в геноме отца этих изменений нет, а у ребенка они появляются. И, что самое интересное, в тех же работах показано: чем старше отец, тем больше в его ДНК поломок передается потомству. К женщинам это, кстати, не относится. В среднем от матери ребенку передается порядка десяти-двадцати замен, но это число не зависит от возраста матери. Повторю еще раз. Смысл в том, что если мы возьмем двух абсолютно здоровых людей и у них родится ребенок, то у малыша будет примерно сорок новых замен от папы и двадцать новых замен от мамы, то есть он будет иметь порядка шестидесяти замен в геноме, которых у родителей нет. И понятно, что эти шестьдесят изменений могу находиться в любом месте. Они могут быть в каких-то важных генах, а могут быть в генах, которые вообще не имеют никакого смысла. Но вероятность того, что изменения эти все же могут повредить какой-то ген и возникнет заболевание аутосомно-рецессивного типа или аутосомно-доминантного, есть всегда. И с этим мы вообще ничего сделать не можем — так устроена природа. И она при этом все-таки довольно совершенна. Вы только представьте: три миллиарда нуклеотидов удваиваются при делении клетки, и это все довольно сложный, большой процесс, и только сорок ошибок могут при этом возникнуть. Заболевания эти всегда были, есть и будут, и человечеству от них не избавиться. Но что самое важное — и это то, чем медицинская генетика занимается, на что направлена, — теперь у нас есть ДНК-диагностика.
Зачем диагностируют генетические заболевания? В первую очередь, для того, чтобы человек понимал свою судьбу. Когда человек чем-то болен, знание и само понимание природы этого заболевания — как оно устроено, почему возникло — психологически сильно облегчают жизнь. Второй и зачастую самый важный момент: ДНК-диагностика нужна для того, чтобы, воспользовавшись знанием, люди могли запланировать рождение здоровых детей. И сейчас я расскажу про то, как это устроено.
Генетических заболеваний много, и причин, по которым они возникают, тоже много. Самые первые исследования этих причин выглядели очень просто: все, что могли сделать ученые 50—60 лет назад, — это посмотреть в микроскоп и увидеть, как устроены хромосомы человека. И сегодня мы знаем, что у человека 23 пары хромосомы. Все возникающие аномалии относительно этой нормы ученые регистрируют, и описывают, и связывают с какими-то генетическими заболеваниями. Вот картинка, в которой несложно разобраться, если внимательно посмотреть, что же такого в ней неправильного.

Легко заметить, что у 21-й хромосомы три копии вместо положенных двух. Не заметить это в микроскоп даже студенту сложно. И понятно, что такое вот аномальное увеличение копий хромосом — как и, наоборот, уменьшение — приводит к заболеванию. То же самое, если хромосома имеет какую-то чрезмерную длину или становится короче. И отсюда возникают хромосомные заболевания. Они представляют собой очень большую группу — около 1% новорожденных имеет такие патологии (и около 2% детей рождается с хромосомными патологиями у женщин старше 35 лет; безусловно — так уж устроена биология — с возрастом некоторые процессы начинают работать хуже, в том числе эмбриональное развитие и многое другое). Вообще статистика хромосомных аномалий очень интересным образом устроена. На 10 тысяч беременностей, которые статистически можно проанализировать, мы имеем порядка 9 тысяч с нормальными хромосомами и порядка 800 случаев с патологиями. И среди этих 800 — вот так уж опять устроена природа — лишь 50 случаев приводят к тому, что рождаются дети, имеющиеся какие-то аномальные вещи. Остальные беременности, как правило, замирают, не развиваются и заканчиваются самопроизвольными выкидышами. С одной стороны, это хорошо. Природа понимает, что должен быть нормальный набор хромосом, а если что-то не так — хромосом становится больше, меньше, теряются какие-то важные фрагменты, — на клеточном уровне запускаются процессы, которые беременность останавливают. С другой стороны, в каких-то случаях беременность продолжается, несмотря ни на что. Один из самых известных примеров — трисомия 21-й хромосомы, которая приводит к синдрому Дауна.
Конечно, на сегодняшний день существуют разные способы всего этого избежать. Микроскопический метод наблюдения за количеством и качеством хромосом, существующий очень давно и очень успешно, потихонечку вытесняется более современным и чувствительным методом, который называется «микроматричный анализ».

Я совсем кратко обрисую, как он устроен: берутся ДНК пациента и контрольная ДНК, хитрым образом готовятся, флуоресцентно метятся и на специальных матрицах гибридизуются, в результате мы видим хромосомы и видим флуоресцентный сигнал, который поступает от разных фрагментов ДНК больного. В каких-то случаях сигнал возрастает, и это означает, что в этом месте возникают копии генов, в каких-то, наоборот, пропадает, и это означает, что какой-то участок в гене был, но пропал. То есть в одном случае возникает дупликация, в другом — делеция. Микроматричный анализ очень чувствительный, и с помощью него происходящие события можно разглядеть с предельно высокой точностью.
Однако основным методом диагностики было и остается секвенирование ДНК. Оно было изобретено в 1980 году замечательным ученым Фредериком Сенгером, который придумал, как можно определять те самые нуклеотиды, из которых состоит наша ДНК. На сегодняшний день такого рода анализ поставлен на поток, его делают практически во всех ДНК-диагностических лабораториях мира. Делается он очень быстро, эффективно, с помощью него можно исследовать отдельные участки генов. Грубо говоря, это основная машина медицинской генетики. Поиск мутаций генов с помощью секвенирования ДНК очень простой: на выходе мы получаем хроматограмму, где за каждым всплеском сигнала стоит какая-то конкретная буква. Когда мы секвенируем ДНК больного, то мы можем обнаружить, что у здорового человека в одном месте буква Т, а у больного в том же месте — буква Г. Найти мутации в каких-то конкретных генах — задача несложная. Главное — понимать, где эти мутации смотреть.

Следующий шаг развития ДНК-диагностики — массовое параллельное секвенирование. Изобретены такие мощные машины, которые могут отсеквенировать ваш геном весь и сразу, то есть все-все хромосомы, все-все гены за один анализ будут проанализированы и расшифрованы. Технология эта появилась относительно недавно и долгое время работала недостаточно качественно. Сегодня все ошибки устранены, и массовое параллельное секвенирование является одним из самых точных и доступных большинству людей анализов. Сделать его может каждый — стоит это порядка 30 тысяч рублей. Сейчас машины для секвенирования выглядят вот так:

Но самое интересное, что они, как ожидают (и правильно делают) ученые, будут в ближайшем будущем вытеснены совсем маленькими, очень компактными мини-секвенаторами, которые будут подключаться к USB-порту. На сегодняшний день эта технология тестируется — пока, правда, делает много ошибок, — но ожидается, что такой аппарат будет стоить всего-то около 200 долларов и будет выглядеть как-то так:

В какой-то мере очень даже хорошо, но в то же время и плохо. Появление таких технологий, способных к подобному роду анализов, привело к тому, что в развитых странах — в частности, в Англии и Нидерландах — бюджетные больницы сделали такой анализ обязательным для всех, кто к ним приходит. Причем неважно, имеет человек генетические заболевания или нет: как только он записался на прием к врачу, ему сразу делают секвенирование генома. Это было введено в 2011 году — то есть относительно недавно, — и сейчас клиник, практикующих такой подход, становится все больше. И все было бы прекрасно и здорово, но на руки врач получает разные буковки — A, Т, Г, Ц, которые могут следовать друг за другом в разном порядке, и количество этих буковок у каждого из нас — порядка трех миллиардов штук.

Самая сложная задача этого подхода (секвенирование генома и его последующий анализ) заключается в том, чтобы расшифровать смысл этой последовательности, интерпретировать ее, понять, где в ней есть мутации, которые вызывают заболевания или предрасположенность к ним, а где их нет. Как только люди научатся понимать эту расшифровку, «читать» эту аннотацию, так сразу начнется следующий этап развития медицинской генетики. Но пока этого не произошло, вряд ли будет полезным, если мини-секвенаторы, так сказать, войдут в наши дома.
Осознание этой проблемы началось вот с этих двух замечательных людей. В 2007 году вышли — параллельно и практически одновременно — две работы, в которых были просеквенированы персональные геномы Крейга Вентера и Джеймса Уотсона (да-да, того самого, который открыл двухцепочечную структуру ДНК, действительно великого ученого; в знак того, что он так много сделал для науки, ему был преподнесен вот такой подарок). В общем, вышли две эти работы по секвенированию индивидуальных геномов человека, в которых ученые пытались вычленить смысл из этой последовательности, и — ничего у них не получилось. Потому что при анализе были найдены гены, отвечающие за конкретный фенотип (цвет глаз, цвет волос) или ответственные за возникновение каких-то заболеваний, но по факту проявлений этих обнаружено не было. Или, наоборот, у Вентера и Уотсона были заболевания и разные физиологические состояния, но ничто в «аннотации» на них не указывало. Возникла пропасть. Вроде знаем буквы — A, Т, Г, Ц, но правильно интерпретировать их смысл не умеем. Пропасть эта имеется до сих пор. Почему? Потому что геном человека очень большой, и если мы просеквенируем ДНК любого из нас, то в результате получим около трех миллионов каких-то индивидуальных различий, которые будут отличать нас друг от друга. Что и было в свое время сделано с Вентером, Уотсоном и другими неизвестными людьми: когда их отсеквенировали, выяснилось, что 3,2 миллиона нуклеотидов отличают Уотсона от всех других. И разобраться, важны ли эти три миллиона и какие особенности важнее других, пока очень и очень сложно. Даже если брать не весь геном, не всю длинную последовательность ДНК, а рассматривать только значимые участки, где содержатся гены, из которых образуются белки, выполняющие какую-то функцию. Таких участков в геноме около одного процента. Суммарно во всех них содержится от 30 до 70 тысяч геномных различий. И разобраться, какие влияют на работу белка, а какие не влияют, с точки зрения медицинской генетики пока что является очень сложной задачей. Работа потихонечку движется, и опять-таки в этом помогает подход, который был обнаружен в самом начале медицинской генетики, — анализ родословной больного. Когда нет нужды сравнивать ДНК данного конкретного человека с ДНК других людей, а можно сравнить с ДНК родственников, то есть генетически близких индивидуумов. Вот ДНК здорового брата, а вот больного — какая разница есть между ними? В этом случае найти причину заболевания становится гораздо легче.
Подобные работы на сегодняшний день как раз являются самыми успешными. И вот одна из них: мальчик болел невропатией, просеквенировали геном родителей и геном мальчика и по разнице того, что есть у родителей, с тем новым, что нашли у ребенка, обнаружили мутацию в гене SLC26A3, и стало понятно, откуда это заболевание, даже были предложены какие-то способы его компенсировать.

99% генетических заболеваний неизлечимы на сегодняшний день. Никак мы пока не можем помочь людям. И то, что медицинская генетика дает, — это лишь предупреждение заболеваний. Я тут составил такую широко известную шараду — «лечить нельзя предупредить», и сейчас мы проговорим все, что подходит под заголовок, который получится из шарады, если запятую поставить после второго слова. То есть — «лечить нельзя, предупредить». И как же предупреждают? Первый способ: ДНК-диагностика носительства мутаций. Вот интересный случай, который описывает, насколько этот метод эффективен и правилен. Есть такое генетическое заболевание Тея—Сакса — очень тяжелое, очень редкое. В возрасте около полугода у детей возникает остановка в психическом и физическом развитии, постепенно теряются зрение, слух, способность глотать, ребенок погибает в возрасте примерно четырех лет. Известно, что мутация, вызывающая это заболевание, находится в гене HEXA, что это аутосомно-рецессивный тип наследования, то есть оба родителя имеют по поломанной копии гена и ребенок наследует обе поломки. Так уж устроено наше существование, что в каких-то странах, которые живут обособленно и закрыто, такого рода заболевания встречаются очень часто. В общем, в случае Тея—Сакса это Израиль. И конкретно евреи-ашкеназы. Один больной ребенок на три тысячи новорожденных. Заболевание тяжелое, а Израиль — это государство, которое заботится о здоровье нации. Потому на государственном уровне было введено обязательное тестирование на носительство мутаций в гене HEXA, и буквально через несколько лет в Израиле стал рождаться лишь один больной ребенок на много сотен тысяч. Похожая история была в Финляндии, которая в последние 300 лет вела себя очень обособленно, не сильно взаимодействуя с миром, и это привело к тому, что отдельные генетические заболевания у финнов стали встречаться очень часто. У них тоже была введена скрининг-программа на носительство сразу нескольких болезней — и за весьма короткий срок их все активнейшим образом фактически элиминировали.

Несколько лет назад в России тоже запустили программу под названием «неонатальный скрининг». То есть, как только рождается ребенок, у него из пятки сразу берут несколько капелек крови и проводят ДНК-диагностику на самые частые в нашей стране генетические заболевания: адреногенитальный синдром, галактоземию, врожденный гипотиреоз, муковисцидоз, фенилкетонурию. Делается это все для того, чтобы на ранних этапах понять, как можно помочь человеку, максимально компенсировать патогенный эффект, не дать ему развиться.

Следующий вариант диагностики — пренатальная диагностика. На ранних сроках беременности аккуратно, не повредив ни плод, ни внутренние органы матери, берут кое-какие части хориона, то есть оболочки плода, по которым делают генетический анализ будущему ребенку, чтобы понять, несет ли он какие-то поломки в своих генах. Если выясняется, что поломки присутствуют, матери предоставляется выбор: прервать беременность или продолжить. Это очень важно — предлагается выбор. Именно так устроено генетическое консультирование: не существует никаких строгих правил, человек сам решает, как ему с этим существовать.

Благодаря появлению мощных секвенаторов теперь есть неинвазивная пренатальная диагностика. Устроена она очень интересно. На ранних сроках беременности — например, на сроке в десять недель — у матери берут кровь из вены. Известно, что в ходе развития плода какие-то его клетки отмирают, ДНК их крошится и попадает в кровоток матери. И если взять у матери кровь и воспользоваться мощным секвенатором, то с помощью специальных алгоритмов можно идентифицировать, какая ДНК материнская, а какая плода, и увидеть, есть ли в геноме плода какие-то замены, мутации. И если мутации есть, то матери опять же предоставляется выбор.

Венцом всех трудов генетиков на сегодня является так называемая преимплантационная генетическая диагностика. Она появилась совсем недавно, является самым сложным, самым трудоемким и самым дорогим диагностическим методом, но позволяет получить здорового на сто процентов ребенка в подавляющем большинстве случаев. Сразу оговорюсь, такая диагностика нужна лишь в тех случаях, когда в лабораторию приходит супружеская пара и говорит: вот у нас первый ребенок родился с таким-то генетическим заболеванием, но мы хотим, чтобы следующий обязательно был здоровым. Генетики проводят анализ всей семейной истории, вычленяют ген с конкретной мутацией и понимают, что нужно делать, чтобы этой мутации у будущего ребенка не было. Как это происходит? У женщины вызывают суперовуляцию, в результате которой получают какое-то количество яйцеклеток. После этого в лабораторных условиях проводится экстракорпоральное оплодотворение. Спустя несколько дней из оплодотворенных яйцеклеток без какого бы то ни было урона для будущего эмбриона отбирают одну-единственную клетку, по которой проводят генетическую диагностику. И если удается получить информацию о том, что у данной оплодотворенной яйцеклетки на стадии бластомера не содержится никаких мутаций, то именно эта яйцеклетка подсаживается матери, которая через девять месяцев даст совершенно здоровое потомство.
Медицинская генетика разработала много разных подходов, позволяющих создать условия, в которых возможно избежать возникновения генетических заболеваний, но, конечно же, в случае новых мутаций, приводящих к возникновению наследственных заболеваний, мы не можем ничего прогнозировать. В результате рождаются больные дети. И понимание того, что их надо лечить (раз уже не смогли предупредить), — это очень и очень актуальная задача. И наука движется и в эту сторону тоже. Главная проблема состоит в том, что речь идет о восьми тысячах заболеваний. Разработать какой-то универсальный подход, который позволил бы в любом случае вылечить все, невозможно даже чисто теоретически. Поэтому для каждого случая генетики изобретают индивидуальные решения, индивидуальные технологии.

Вот здесь приведена диаграмма, на которой указаны самые разные терапевтические стратегии, применяющиеся для попыток лечения генетических заболеваний обмена веществ. Видно, что в каких-то случаях применяют хирургию, в каких-то — тканевую трансплантацию или трансплантацию костного мозга. Один процент занимает генная терапия, когда осуществляется доставка здоровой копии гена. В каких-то случаях пытаются ограничивать болезнь диетой или приемом лекарственных препаратов. В общем, подходов много.
Что касается диеты, то в некоторых случаях она устраняет заболевание практически полностью. Два широко известных заболевания — галактоземия и фенилкетонурия. Первое — это нарушение углеводного обмена, когда фермент, который усваивает молочный сахар, мутировав, перестает его расщеплять. Но когда рождается ребенок, он, понятно, питается исключительно материнским молоком. И в этом случае неусваивание молока потихонечку приводит к тому, что начинают развиваться разные патологии — в частности, желудочно-кишечные проблемы, цирроз печени, катаракты. Все это происходит буквально в течение первых недель и, к сожалению, довольно часто приводит к летальному исходу. Тем временем всего лишь посредством удаления молока из рациона ребенка можно сделать так, что это врожденное генетическое заболевание не будет проявляться. То же самое с фенилкетонурией — заболеванием, которое входит в пренатальный скрининг в России. Фермент, ответственный за метаболизм аминокислоты фенилаланина, мутировав, перестает работать. Но если убрать из пищи продукты, которые содержат этот самый фенилаланин (в том числе некоторые орехи, грибы, некоторые молочные продукты), ребенок будет развиваться здоровым. Однако таких историй про диеты совсем немного, по пальцам перечесть. Ученые пытаются для каждого больного подобрать питание, чтобы как-то облегчить его существование. Но все заболевания разные, и мутации не всегда приводят к тому, что ген сломался и больше не работает. Иногда функция белка сломана частично и он работает не так эффективно, поэтому достаточно что-то где-то слегка компенсировать — и эффект значительный.
Вероятно, что именно в Китае будет сделан — если уже не сделан кулуарно, что тоже все обсуждают, — «отредактированный» человек.
В целом способы терапии наследственных заболеваний можно разбить на две группы. Первая — за счет низкомолекулярных соединений, когда можно подобрать какую-то таблетку, которая скомпенсирует существующие поломки. Несмотря на то что мы все привыкли пить таблетки, в случае с наследственными заболеваниями это очень редко работает. А вот то, что должно чисто теоретически всегда работать — и в случае с генетическими поломками в первую очередь, — это использование разных молекулярных методов, куда сейчас и движется вся медицинская генетика. Самое интересное, что для того, чтобы использовать все эти подходы, существует целый арсенал разных вариантов. Есть методы, которые позволяют активировать работу каких-то конкретных генов: ген не работает, и мы можем сделать так, чтобы он в данной клетке активно заработал. Или, наоборот, работает мутантный ген, производит токсичные продукты, но существуют подходы, которые могут подавить его работу селективно, направленно, чтобы именно он среди всего генного многообразия перестал работать.
Самое последнее и интересное, что родилось в генетике буквально пять лет назад, — это редактирование мутантной копии гена. Подходы, которые позволяют исправить мутацию. Я расскажу об этом и о том, куда это все движется, но сперва напомню постулат, который всем нам, по идее, известен, — центральную догму молекулярной биологии. Помните, я рассказывал о великом ученом Джеймсе Уотсоне? Так вот, у него был друг — Фрэнсис Крик, с которым они вместе открыли двухцепочечную структуру ДНК. Потом Уотсон стал заниматься одними вещами, а Крик — другими (хотя оба работали в области молекулярной биологии). Но так или иначе именно Крик сформировал на основе структуры молекулы ДНК эту самую центральную догму: у нас есть ДНК, с нее получается РНК, с нее получается белок, являющийся венцом всей этой истории, который дальше как-то функционирует, выполняет какие-то функции. На сегодняшний день описано огромное множество белков, объяснено, как они устроены, какие у них есть части, отвечающие за функционирование. Для чего все это описывается? Для того, чтобы потом построить вот такую огромную генную сеть.


Мы понимаем, какие процессы с какими генами связаны, как эти процессы запускаются, как передаются сигналы из одного места в другое, как они регулируется. Мы понимаем, как применять все эти подходы, чтобы какой-то ген выключить, а какой-то включить. То есть, если возникла мутация в гене, это не значит, что все, конец, мы не можем ничего сделать. Нет, мы можем понять, какой обходной путь активировать, чтобы наладить нормальное существование клетки, нормальный метаболизм. Или — наоборот — какой путь подавить. И фундаментальная наука здесь очень нужна для того, чтобы эти знания дальше можно было использовать в генетике для лечения наследственных заболеваний. Но почему так сложно бывает применять эти подходы? Потому что человек — это многоклеточный организм. То, каким вы меня сейчас видите, — это миллиарды клеток, у каждой своя программа, и все занимаются какими-то своими функциями. В мозгу одни клетки, в мышцах другие. И это все очень сложно устроено, поэтому, если я намерен вмешаться в какой-то процесс, мне будет необходимо добраться не до всего организма в целом, а именно до тех клеток, которые реализуют конкретный фенотип. Если больной страдает от судорог, значит, в нейронах происходит плохая передача сигналов, и мне нет необходимости вмешиваться в работу всех клеток, мне нужно добраться до клеток мозга. Соответственно адресная доставка — самая сложная задача, стоящая сегодня перед учеными. Она легко реализуется, когда есть какой-то конкретный орган, хорошо изолированный, куда можно все доставить без каких-либо проблем. Самый простой пример — это глаз. Так уж заведено природой, что вот он, такой доступный, такой изолированный — и делай с ним что угодно. И довольно много чего с ним уже делают — в том числе применяют генную терапию, когда можно какие-то процессы подкрутить или, наоборот, подавить. Но есть и другой хорошо доступный орган — кожа. Казалось бы, вот она, но — не получается доставить туда материалы, потому что кожа имеет защитный барьер, не позволяющий, чтобы туда попадала всякая ерунда.

Есть, к примеру, такое заболевание — миодистрофия Дюшенна, очень распространенное. Чтобы его лечить, ученые разрабатывают разного рода подходы. И видите, тут не один подход, над которым ученые работают в течение многих лет, а много подходов. Где-то пробуют клеточную терапию — больному доставляют новые клетки, чтобы понять, будет ему лучше или не будет. Где-то добавляют разные лекарственные соединения. У кого-то получается лучше, у кого-то не получается совсем. Один из последних вариантов: ученые разработали специальное химическое вещество, которое позволяет избегать мутаций, вызывающих появление так называемых стоп-кодонов. Что это такое? В каких-то случаях мутация приводит к тому, что рибосома, когда двигается по РНК и синтезирует белок, доходит до возникающего за счет мутации стоп-кодона, и в результате получается укороченный белок вместо нормального, большого. Укороченный белок не может нормально функционировать. И ученые придумали такое вещество, которое помогает рибосоме при движении по молекуле РНК перестать распознавать эти стоп-кодоны, в результате получается длинный продукт. Это, на самом деле, очень большое достижение — и произошло оно совсем недавно.
Однако, несмотря ни на что, основная история, на которую все очень рассчитывают, — это генная терапия. Когда мы можем копию гена, которая не несет никаких мутаций, каким-то образом доставить в клетку с мутантным геном. Для этого существует так называемый вирусный способ доставки. То есть те самые вирусы, от которых мы болеем, специальным образом модифицируют, убирают все ненужные компоненты, оставляют только структурную часть и используют как транспорт для доставки нормального гена человека. Идея эта придумана очень давно, и сегодня ее пытаются реализовать во многих странах одновременно. Несколько тысяч испытаний сейчас происходит во всем мире.
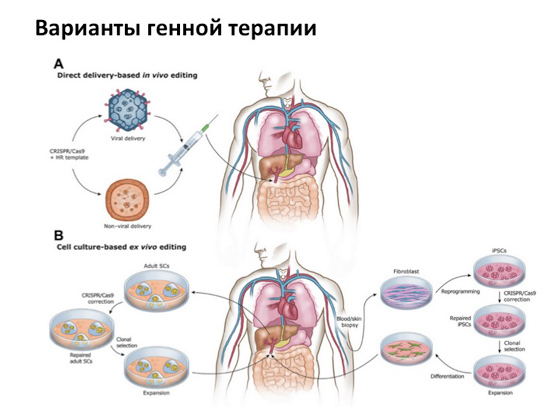
Под конец я вам расскажу несколько удачных историй. Первая связана с заболеванием, которое называется синдромом дефицита аденозиндезаминазы. Возможно, вы слышали о фильме «Мальчик из пузыря» (или даже смотрели его) — про то, как жил-был мальчик, все время находился в пузыре, мама его ото всего оберегала, а он сбежал из дома и увидел, какой мир прекрасный. Эта история основана на реальных событиях. Мальчика звали Дэвид Веттер, у него было именно такое генетическое заболевание, связанное, по сути, с тяжелой формой иммунодефицита. Грубо говоря, ребенок мог погибнуть от любой инфекции. И когда стало понятно, что он так сильно болен, его изолировали от всего внешнего мира в комнатах, где он прожил довольно долго. Его все время пытались лечить разными методами, разными препаратами. У него была сестра, от которой ему делали пересадку костного мозга, — но и это не помогло. История, в общем, грустная: мальчик умер, не дожив немного до того момента, когда генная терапия была успешно применена к лечению этого заболевания. Выглядит это следующим образом: от больного берут клетки костного мозга, которые отвечают за производство иммунных клеток, с помощью вируса доставляют в них копию гена и затем клетки подсаживают обратно больному. В результате человек получает новый, так скажем, костный мозг с копией нормального гена. История эта была впервые реализована в 1990 году.
Еще одна история касается врожденного амавроза Лебера — это дегенерация сетчатки глаза, довольно редко встречающаяся и возникающая из-за мутации в определенном гене, кодирующем белок, который отвечает за пигментный эпителий сетчатки. Были разработаны специальные вирусные частицы, в которых была нормальная копия гена RPE65, и больным, страдающим этим заболеванием, были сделали инъекции препарата прямо в глаз. В результате больные, потерявшие зрение или даже с рождения его не имевшие, вновь его обрели при лечении. В прошлом году этот генный препарат был одобрен специальным комитетом FDA, и его должны вот-вот запустить в массовое использование.
Таких историй немало, но наследственных заболеваний, как вы помните, очень много, около восьми тысяч.

Под конец, конечно, мне очень хочется хоть пару слов сказать о такой замечательной технологии, как редактирование генома. Она произвела полнейший фурор в науке. Суть этого редактирования в следующем: есть определенного рода белок, который обладает нуклеазной активностью, то есть может расщеплять ДНК, а за счет одной хитрой РНК мы можем программировать работу белка так, чтобы он разрывал ДНК в каком-то конкретном месте. А дальше возможны два варианта. Для нас сейчас важен один из них: когда белок разорвет ДНК в заранее запланированном месте, содержащем мутацию, можно активировать механизмы клеточной репарации, позволяющие разрыв восстановить так, что возникнет новая копия гена, которая не будет нести мутацию. И создание такого рода белков, которые специфично могут быть куда-то направлены, в какие-то определенные гены, — это очень важный шаг. И при этом очень простой и удобный метод: любой студент в хорошо оснащенной лаборатории может с этим справиться за считанные месяцы. Поэтому сейчас выходит много публикаций о том, где и когда эту технологию применяют, и понятно, что в первую очередь ее пытаются применить для лечения разных генетических заболеваний. И пока, судя по научным статьям, все идет хорошо. Я читал недавно статью в журнале Nature, в которой анализировалось, как в разных странах на сегодняшний день устроены законодательные акты и где впервые должен появиться «отредактированный» человек — неважно, с суперспособностями или просто вылеченный от какого-то заболевания. Если не вдаваться в подробности, то абсолютно все страны это не поддерживают. Но вопрос в том, насколько это глубоко проговорено на законодательном уровне. В общем, в статье говорится, что одна из потенциальных стран, в которой все эти моменты проговорены менее четко, — это Китай. Причем если раньше считалось, что Китай — отсталая в научном плане страна, то сейчас они нас обогнали так, что их уже и не догнать, особенно в биологии. Китайцы вкладывают в это огромное количество денег. Они привлекают специалистов со всего мира, своих посылают обучаться, а потом берут обратно и дают им лаборатории, институты. Они очень активно в этом направлении развиваются. Вероятно, именно в Китае будет сделан — если уже не сделан кулуарно, что тоже все обсуждают, — «отредактированный» человек. И самая первая история, которую долго не могли опубликовать, была про то, что уже демонстрировали метод редактирования генома на человеческих эмбрионах. Ничего страшно там не было, все очень хорошо и правильно было сделано, эмбрионы человека не могли размножаться дальше и были взяты как модельный объект. В общем, эксперимент показал, что при редактировании пока возникает много ошибок. Как это принято в научном сообществе, любая новость такого рода самым активным образом дискутируется: с одной стороны, все понимают — да, все эти технологии, что только что появились, годятся для решения разных задач. С другой стороны, побочные эффекты, которые могут появиться, никто не может предсказать. И потому ученые очень аккуратно, шаг за шагом делают попытки все это проверить и исследовать. Я очень не хочу зря обнадеживать, но, как мне кажется, в ближайшие десять-двадцать лет у нас точно появятся надежные средства для лечения с помощью генной терапии. А пока имеем то, что имеем.
Записала Наталья Кострова
 Поцелуй Санта-Клауса
Поцелуй Санта-Клауса
Запрещенный рождественский хит и другие праздничные песни в специальном тесте и плейлисте COLTA.RU
11 марта 2022
14:52COLTA.RU заблокирована в России
3 марта 2022
17:48«Дождь» временно прекращает вещание
17:18Союз журналистов Карелии пожаловался на Роскомнадзор в Генпрокуратуру
16:32Сергей Абашин вышел из Ассоциации этнологов и антропологов России
15:36Генпрокуратура назвала экстремизмом участие в антивоенных митингах
Все новостиЛитература Colta Specials
Colta SpecialsПоэтесса Наста Манцевич восстанавливает следы семейного и государственного насилия, пытаясь понять, как преодолеть общую немоту
20 января 20226121 Искусство
Искусство Искусство
Искусство Молодая Россия
Молодая РоссияРассказ Алексея Николаева о радикальном дополнении для обработки фотографий будущего
18 января 20222284 Литература
Литература Общество
Общество Искусство
ИскусствоКуратор Алиса Багдонайте об итогах международной конференции в Выксе, местном контексте и новой арт-резиденции
17 января 20225782 Академическая музыка
Академическая музыка Искусство
Искусство Литература
Литература Общество
Общество
Андрей Мирошниченко о недавнем медиаскандале, который иллюстрирует борьбу старых и новых медиа
13 января 20229160